In the intricate world of laboratory analysis, two stalwarts, the microplate reader and the spectrophotometer, reign supreme. These instruments share a common measurement principle, utilizing the Lambert-Beer law to measure sample absorbance. This article aims to shed light on differences between microplate reader and the spectrophotometer, focusing on three key aspects.

1. Major Differences
(1) Container for the Solution:
Microplate Reader:
Microplate readers utilize a plastic microplate, usually made of transparent polyethylene.
Each microplate can hold multiple solutions, often in 48 or 96 wells.
Requires a special photoelectric colorimeter due to unique requirements.
Spectrophotometer:
Employs a cuvette to contain the solution.
Each cuvette holds a single solution at a time.
Provides a straightforward container without additional solid-phase carrier properties.
(2) Direction of the Light Path:
Microplate Reader:
Features a vertical light path.
Light passes through the microplate vertically, either from top to bottom or vice versa.
The vertical light path minimizes the impact of liquid concentration or dilution.
Spectrophotometer:
Adopts a horizontal light path.
The light path remains constant regardless of the container's properties.
Affected by sample liquid level, microplate plate's light transmittance, and well bottom flatness.
(3) Length of the Optical Path:
Microplate Reader:
Vertical light path necessitates the length of the optical path to be the height of the liquid surface.
Measurement influenced by sample volume due to the unique path length.
Spectrophotometer:
Spectrophotometer adopts a fixed optical path length of 1 cm in the cuvette.
Ensures comparability across instruments and batches.

2. Advantages and Disadvantages Comparison
Spectrophotometer:
Advantages:
Wide detection wavelength range.
Consistent sample additions do not affect results.
Disadvantages:
Heavy workload, cumbersome operation, and time-consuming.
High reagent consumption.
Poor result stability, repeatability, and potential for large errors.
Difficulty in detecting and tracing substances.
Main Application Areas:
Qualitative and quantitative analysis in drug inspection, drug analysis, environmental testing, health, epidemic prevention, food, chemical industry, and scientific research.
Microplate Reader:
Advantages:
Processes a large number of samples simultaneously, saving time.
Requires fewer samples and reagents.
Simple operation, good repeatability, fast detection, and high efficiency.
Disadvantages:
96-well plate strong UV absorption below 300nm, limiting use for content determination.
Requires precise sample volume consistency, demanding skilled operation.
Main Application Areas:
Clinical testing, biological research, agricultural science, food, and environmental science.
In conclusion, understanding the distinctions between microplate readers and spectrophotometers is crucial for selecting the right tool for specific laboratory applications. Each instrument brings unique advantages and limitations, contributing to their diverse roles in scientific research and analysis.
X-ray diffraction (XRD) is a commonly used testing method, yet many students remain unfamiliar with its principles and applications. In this article, Drawell will provide insights into XRD, addressing various aspects of this technique.
1. Understanding the Utility of XRD
X-ray diffraction involves scattering X-rays when they interact with a material. When X-rays strike crystalline substances, they undergo coherent scattering, which is a diffraction phenomenon. This means that the incident X-ray beam changes direction without changing its wavelength upon exiting the material. This unique behavior is specific to crystalline substances.
Most solid-state materials are either crystalline, microcrystalline, or quasi-crystalline and can exhibit X-ray diffraction. The crystal's microstructure exhibits a periodic, long-range ordered arrangement. The X-ray diffraction pattern is a representation of the three-dimensional structure of the crystal microstructure, containing critical information about the crystal's composition and structure. XRD is currently the most powerful method for studying crystal structures, including atomic positions, unit cell dimensions, and more.

XRD is particularly well-suited for phase analysis of crystalline substances. Different phases or structures within crystalline substances exhibit variations in the number of diffraction peaks, their angles, relative intensities, and peak shapes. By comparing the X-ray diffraction pattern of an unknown sample with that of known crystalline substances, one can qualitatively identify the phase composition and structure of the sample. Additionally, quantitative analysis of the sample's phase composition is possible by analyzing the diffraction intensity data.
2. Distinguishing Amorphous, Quasi-Crystalline, and Crystalline Structures
Differentiating between amorphous, quasi-crystalline, and crystalline structures in XRD patterns is not always straightforward. In XRD patterns obtained from crystalline materials, you typically observe sharp, distinct peaks with narrow 2Θ widths at half-height (usually 0.1° to 0.2°). Broadened peaks suggest that the crystals in the sample have smaller particle sizes, often less than 300 nm, known as "microcrystals." The Scherrer formula can estimate the grain size based on spectral line broadening.
In contrast, amorphous materials exhibit a gentle, continuous change in X-ray scattering intensity over a wide angle range (2θ 1° to several tens of degrees), often with one or more maxima. This phenomenon occurs due to extremely fine grain sizes leading to broadened, overlapping, and blurred diffraction peaks. Quasi-crystalline materials represent a transitional state between crystalline and amorphous, characterized by unique diffraction patterns.

3. Impact of Different X-ray Targets
The choice of X-ray target, such as copper or chromium, affects the characteristic wavelengths used in XRD experiments. The diffraction angle, determined by the Bragg equation, changes with varying wavelengths. Consequently, the positions of diffraction peaks on XRD patterns obtained from different X-ray tubes will differ systematically. However, a crystal's set of d values, inherent to its structure, remains consistent, independent of the X-ray target.
The relative intensities of diffraction peaks may vary slightly when using different targets for the same sample. This variation is attributed to the absorption properties of the sample in relation to the incident X-ray wavelength and the target material.
4. Determining Crystal Planes Corresponding to Diffraction Angles
To determine the crystal planes corresponding to different diffraction angles, consult powder diffraction data cards that provide diffraction index information for each diffraction line. For unknown crystal structures, the process of determining diffraction indices for each line is known as "indexing the diffraction pattern." This step requires a foundation in crystallography and proficiency in indexing software, such as treaor90.
5. Obtaining Atomic Coordinates in Crystals
To acquire atomic coordinates within a crystal, perform single-crystal X-ray diffraction. This technique, in addition to using CCD detectors, provides precise atomic position data.
6. Calculating Grain Size, Lattice Constants, and Distortion
Grain size, lattice constants, and distortion can be calculated from X-ray diffraction data, specifically from peak shape information. When the broadening of diffraction peaks is solely due to crystal grain fineness, the Scherrer formula can estimate grain size based on the extent of peak broadening.
The above is the arrangement of the common problems of XRD. If you need an XRD test or the XRD instrument, you can contact Drawell.
Introduction:
Raman spectrometers, also known as Raman Spectrum Analyzers, have emerged as powerful tools for molecular structure determination. Leveraging the phenomenon of Raman shift, these devices enable both quantitative and qualitative analysis across a wide range of samples, including solids, liquids, gases, organic compounds, and polymers. With features like 100 SERS libraries, 3000 constant substance libraries, and cutting-edge technology, Raman spectrometers play a pivotal role in swiftly and accurately detecting illicit additives, chemical contaminants, and adulterants in everyday food items. This article explores the various facets of Raman spectrometers, their types, components, working principles, and diverse applications.

Types of Raman Spectrometers:
Presently, Raman spectrometers come in several varieties tailored to specific applications. These include Fourier Transform Raman spectrometers, confocal microscopic Raman spectrometers, and surface-enhanced laser Raman spectrometers.
Components of Raman Spectrometers:
While the specific composition of Raman spectrometers may vary, they typically consist of essential components that include a laser light source, sample device, optical filter, monochromator (or interferometer), and detector. Each component plays a crucial role in the instrument's functionality.
Optical Filter:
The laser's scattered light, or Rayleigh light, is significantly stronger than the Raman signal and must be filtered out before reaching the detector. Furthermore, to prevent external radiation from interfering with the sample, suitable filters or physical barriers are incorporated.
Monochromator and Michelson Interferometer:
These components come in various configurations such as single grating, double grating, or triple grating. The planar holographic grating interferometer, akin to those used in FTIR, is frequently employed. Various types of beam splitters, including multi-layer silicon-coated CaF2 and Fe2O3-coated CaF2, as well as quartz and extended-range KBr beamsplitters, are available.
Detector:
Traditionally, photomultiplier tubes were used, but modern Raman spectrometers primarily employ CCD detectors. Common detectors for FT-Raman include Ge and InGaAs detectors.
Excitation Light Source:
Several excitation light sources are utilized, including Ar ion lasers, Kr ion lasers, He-Ne lasers, Nd-YAG lasers, and diode lasers. These sources emit wavelengths ranging from 325nm (UV) to 1064nm (IR).
Sample Device:
Raman spectrometers provide various sample placement options, including direct optical interfaces, microscopes, fiber optic probes, and specialized sample holders.

Principle of Raman Spectrometers:
The functioning of Raman spectrometers, also known as the Raman spectroscopy principle, hinges on the concept of Raman shift. This shift refers to the frequency difference between scattered light and incident light, which is solely dependent on the molecular structure of the scattering substance. Raman scattering occurs due to changes in molecular polarizability, with Raman shift reflecting alterations in molecular vibrational energy levels. Different chemical bonds or groups possess characteristic molecular vibrations, leading to distinctive Raman shifts. This principle underpins the qualitative analysis of molecular structures using Raman spectroscopy.
Applications of Raman Spectrometers:
Raman spectrometers find widespread applications across diverse industries, including:
Biology: For detecting low concentrations of biohazardous substances and quantitatively assessing algal lipid content.
Forensic Authentication: In the identification of drug grids, explosives, fibers, hair, pigments, inks, and cyanotoxin solvents for fiber fabrics.
Homeland Security and Defense: For explosives detection, unknown substance identification, border patrol, and security inspections.
Geology: Enabling non-destructive identification of geological materials, gem certification, and origin determination of minerals and gems.
Pharmacy: In drug isoform/solvent detection, drug crystal identification, content analysis of pharmaceuticals, and quality control of ingredients.
Chemistry: For monitoring and confirming input/output substances, process analytical technology (PAT), and identifying resins, petrochemicals, and chemical crystals.
Food Safety and Agriculture: In inspections at ports of entry, pesticide and herbicide assessments, field audits, and bacterial contamination detection.
Semiconductors and Thin Films: For defect inspection of wafers, thin film coating, and quality control in the junction process.

Conclusion:
Raman spectrometers have revolutionized molecular analysis due to their versatility, accuracy, and non-destructive nature. With their ability to rapidly detect molecular structures and various contaminants, they have become indispensable tools in ensuring product safety, quality, and security across diverse fields.
Raman spectrometers are indispensable tools in laboratory testing. As a Raman spectrometer manufacturer, Drawell is here to address some of the most frequently asked questions about these instruments.
1. Laser Raman Spectroscopy vs. Infrared Spectroscopy
To grasp the difference between laser Raman spectroscopy and infrared spectroscopy, imagine their spectral shapes. Infrared spectra appear "concave," while Raman spectra are "convex." These two techniques complement each other in several ways:
Both are vibrational spectra, measuring the excitation or absorption of the ground state with the same energy range.
Raman is a differential spectrum. Infrared is like buying a Coke for $0.01 – straightforward. Raman, on the other hand, is like investing $1 and getting a Coke plus 90 cents back, but you still know the Coke's price.
They follow different selectivity rules. Infrared measures changes in molecular dipole moments, while Raman detects changes in molecular polarizability.
Infrared is known for its strong and easily measurable signal, while Raman typically has a weaker signal.They use different wavelength ranges. Infrared relies on mid-infrared light, which can't penetrate many optical materials. Raman, however, offers various wavelength options, from visible light to near-infrared.
Sample preparation for infrared can be complex and time-consuming, potentially damaging the sample. Raman spectroscopy doesn't face these challenges.
In most cases, Raman and infrared spectroscopy complement each other, with one being strong where the other is weak.2. Blue Shift and Red Shift
Blue shift and red shift describe shifts in wavelength or wave number:
Redshift means a wavelength moves toward longer wavelengths and lower frequencies, often seen in astronomical observations.
Blueshift refers to a shift toward shorter wavelengths and higher frequencies.
These shifts can also occur in molecular spectroscopy, affecting the position of absorption peaks in chromophores due to factors like molecular interactions and solvents.
3. Laser Light Sources in Raman Spectrometers
Raman spectrometers use various laser light sources:
Argon ion lasers
Semiconductors
Helium-neon lasers
Solid-state diode-pumped lasers
Near-infrared lasers (e.g., 785nm)
Neodymium-doped yttrium aluminum garnet (YAG) lasers (1064nm)
Dye lasers
The choice depends on your research object and the need to avoid interference, such as fluorescence.

4. Sample Pretreatment for Laser Raman Testing
Sample pretreatment in laser Raman testing is generally straightforward. Solids and liquids typically require no pretreatment, while gases can be more challenging, especially if they have low density. Polishing the sample surface or cleaning with solvents like alcohol or acetone is often sufficient.
5. Choosing the Excitation Wavelength
The excitation wavelength choice in Raman spectroscopy depends on whether the sample fluoresces under laser excitation. If fluorescence interferes, a different laser should be used. Shorter excitation wavelengths are preferred theoretically, but practical limitations, such as detector sensitivity, can impact the choice.
6. Fourier Transform Raman Spectroscopy vs. Laser Raman Spectroscopy
Fourier Transform Raman Spectroscopy uses a near-infrared laser (1064nm) for organic sample analysis with a weak signal.
Laser Raman Spectroscopy uses lasers of different wavelengths (200-800nm) for high-energy, high-sensitivity measurements.
Fourier Raman can reduce fluorescence interference.
It is generally more affordable.
Dispersion laser Raman is more popular among users.
In conclusion, these FAQs provide insights into Raman spectrometers, but there's more to explore. If you seek further information or wish to find the right Raman spectrometer for your needs, please don't hesitate to contact us. We'll guide you to the ideal product.
In the realm of analyzing test substances, the preliminary treatment of samples presents a complex challenge. This aspect is pivotal within the scope of analysis and detection efforts, constituting a significant facet for identifying the origins of detection inaccuracies. Consequently, this article aims to synthesize four fundamental sample pretreatment techniques employed in detection endeavors of Atomic Absorption Spectrophotometer (AAS) . Additionally, it delves into the foundational detection techniques for six categories of routine samples. These methodologies offer practicality and serve as valuable references for users of Atomic Absorption Spectrophotometers (AAS).

The Four Core Techniques for General Sample Pretreatment in AAS
Wet Digestion Method
For samples approximately ranging from 0.1000 to 0.5000g, prevalent mixed acids are utilized, with varying ratios as follows:
HNO3 : HClO3 = 5 : 1
HNO3 : H2SO4 = 5 : 1
HNO3 : HCl = 5 : 1
Pure HNO3
Note: Volatile (such as acetone, ether, ethanol, etc.) and flammable, explosive substances must be absent during the digestion process. While the wet digestion method is widely employed, its operational specifics will not be reiterated here.
Dry Ashing Method
Suitable for sample sizes typically ranging from 2.000 to 5.000g, this method involves careful treatment to prevent volatilization. The process entails placing the sample in a porcelain crucible, moistening it with a few drops of water, adding a small quantity of concentrated nitric acid, heating it gently to carbonize, and subsequently ashing it in a muffle furnace at around 550°C for 2 to 4 hours. Post-cooling, the residue is dissolved with additional acids plus HNO3 (1:1), which varies depending on the sample. The solution is then filtered, adjusted to volume, and readied in 10mL, 25mL, and 50mL portions for subsequent use.
High-Pressure Vessel Method (Utilizing Polytetrafluoroethylene Lid-Made Tanks)
When dealing with samples below 0.3000g, this method involves introducing 6mL of mixed acid and 1mL of HF (H2O2) to the sample. The lidded autoclave is then sealed, and heating is performed at 160°C for 5 hours. After cooling, the sample is filtered, adjusted to volume, and set aside for future use.
Microwave Digestion Method
In this approach, commonly employed mixed acids encompass:
HNO3 : HClO3
HNO3 : H2SO4
Pure HNO3
It is essential to select the appropriate acid for specific digestion depending on the sample. Readers are encouraged to make suitable choices as per their requirements.
Conventional AAS Analysis and Detection Approaches for Diverse Samples
Analysis of Pb, Cd, As, Mo, Cr, etc. (Graphite Furnace AAS Method)
Pb: Extract 1.0mL of the sample, dilute it to 10mL with 1% HNO3. Linear range: 0~20ng/mL, drying temperature: 80~100°C, ashing temperature: 200°C, atomization temperature: 1500°C.
Cd: Extract 1.0mL of the sample, dilute it to 10mL with deionized water. Linear range: 0.1~0.4ng/mL, drying temperature: 80~100°C, ashing temperature: 200°C, atomization temperature: 1800°C.
As: Extract 1.0mL of the sample, add 100μL of Ni (2mg/mL), dilute to 10mL with 1% HNO3. Linear range: 0~4ng/mL, drying temperature: 80~100°C, ashing temperature: 200°C, atomization temperature: 2200°C.
Mo: Prepare a 1.0mL sample diluted with 1% HNO3 to 10mL. Using Pd as a modifier, the linear range is 0-20ng/mL. Drying temperature: 80-100°C, ashing temperature: 1200°C, atomization temperature: 2600°C.
Cr: Sample 1mL, dilute to 100mL with deionized water, and proceed with appropriate testing. Linear range: 0~40ng/mL, drying temperature: 80~100°C, ashing temperature: 1000°C, atomization temperature: 2600°C.
Analysis and Testing of Se, K, Na, Ca, Mg, etc. in Plant Samples
Plant samples are subjected to washing, drying, fixation at 150°C for 10 minutes, subsequent drying at 70°C, and final comminution before analysis.
Se in forage grass: Extract 1mL of the sample, add 100μL of Ni(NO3)2 improver (3mg/mL), dilute to 10mL with 1% HNO3 and 0.1% Triton (1:1). Linear range: 0~20ng/mL, drying temperature: 80~100°C, ashing temperature: 1000°C, atomization temperature: 2400°C.
K and Na in plants: Weigh 0.2000g of the sample, add 10mL of mixed acid, extract in a constant-temperature water bath at 90°C, filter to 50mL volume, and proceed with suitable sampling and testing.
Ca and Mg in plants: Incinerate the sample at 550°C, dissolve with hydrochloric acid (1+1), adjust to 50mL volume (with 5% CsCl2 modifier 2 mL), and proceed with appropriate sampling and testing.
Beryllium Analysis in Mushrooms and Tea (Transverse Heating AAS Method)
Prepare the sample by washing, drying, fixation at 105°C, subsequent drying at 70°C, cooling, and crushing through a 60 mesh sieve.
Weigh 1.0000g of sample, add 15mL of HNO3, 1mL of H2SO4, digest at a low temperature, dilute to 25mL, and proceed with suitable sa
mpling for testing. Linear range for Be: 0~8ng/mL. Test conditions: drying temperature: 130°C, ashing temperature: 1500°C, atomization temperature: 2300°C.
Germanium Testing in Beverages (Utilizing Transversely Heated Flat Graphite Tubes)
Test outcomes indicate a linear range of 0~200ng/mL, using Ni(NO3)2 improver (50μg/mL) in a 1% HNO3 medium. Instrument conditions: drying temperature: 130°C, ashing temperature: 800°C, atomization temperature: 2000°C.
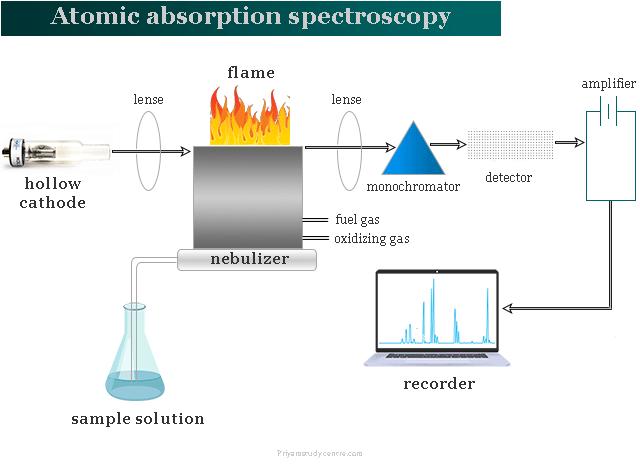
Calcium Detection in Steel Slag (Typically Employing Flame AAS Method)
For samples above 200 mesh, weigh 0.0500g into a 50mL PTFE crucible. Dissolve it with a water:nitric acid:hydrofluoric acid ratio of 5:8:6. Add sulfuric acid, heat, and adjust volume for analysis. To mitigate niobium interference, include
Triton-100 (20%), Vc (0.1 M/L), and 2% HNO3 as improvers.
Determination of Metal Elements in Blood Samples
Human blood encompasses diverse trace constituents, including heavy metal elements. Various detection methods can discern these components, with AAS being particularly effective. Specific digestion methods are elaborated in sections "2, 3), (3)." For instance:
Cu determination in serum (flame AAS method): Dilute 0.8mL of serum with 1% HNO3 to 10mL for testing.
As determination in serum (graphite furnace AAS method): Dilute with 1% HNO3 and utilize Ni(NO3)2 as a matrix modifier.
Cd determination in serum (graphite furnace AAS method): Process 2mL of serum with HNO3 and H2O2 for analysis.
Fe and Cu determination in albumin (flame AAS method): Utilize 1mL of the sample with mixed acid, adjust volume, and proceed with appropriate sampling.
Germanium determination in whole blood (graphite furnace AAS method): Dilute 1.0mL of venous blood, utilize a matrix modifier mixture, and perform analysis at defined temperatures.
This comprehensive overview highlights essential techniques for accurate analysis and detection of various samples by using atomic absorption spectrometry.